INTRODUCTION
Photodynamic therapy (PDT) is an elegant light based oncologic intervention. As currently practiced, a photosensitizer (PS) is applied then activated by light of the appropriate wavelength and intensity. This creates the photodynamic reaction (PDR) which is tumor and vascular ablative.1 This paper will review the mechanisms of action that allow PDT to be a reliable oncologic therapy. We will also offer insight to clinicians and scientists on ways and means to improve therapy and outcome. Following a brief historical review, we will detail the components of PDT that will then allow us to better understand the mechanisms of action of this underutilized cancer treatment.
HISTORY
PDT was accidently discovered over 100 years ago by medical student Oscar Raab.2 He was studying the interaction of fluorescent dyes on infusaria. Raab found that intense light applied to the dye resulted in rapid destruction of these microorganisms. This new light based therapy was more formally described and elucidated by Raab's professors Jesionek and von Tappeiner who coined the ablative process, Photodynamische Wirkung, best translated as The PDR, and PDT was born. By the early 1900's, patients were being successfully treated by this process for a wide variety of cancers, particularly of the skin. Despite this early success, PDT did not achieve enough momentum and was lost for nearly 50 years when the PDR was rediscovered by Lipson and Schwartz. Studies during the 1950's to 1960 revealed not only tumor ablation but the inter-related ability of photosensitizing agents to fluoresce and demarcate tumors.3,4
However, it was not until the 1970's when Dougherty,5 working with porphyrin compounds, accidentally rediscovered PDT. In contrast to previous iterations, Dougherty created a commercially suitable photosensitizing drug, reliable light sources and appropriate clinical trials proving the value of PDT to the oncologic community. For this he is affectionately known as "The Father of PDT," though many other important figures helped bring PDT to a worldwide audience.
PDT COMPONENTS
To better understand the mechanisms of action of PDT, its components need to be defined and explored. Fundamentally, a PS agent is introduced, and then activated by light. When in the presence of oxygen, this active PS may create the PDR. This occurs in a complex interplay of time and space which should ultimately result in lesion ablation and sparing of normal tissue. We will look at each component separately, with a focus on how each effects PDT's mechanistics.
PHOTOSENSITIZING AGENTS
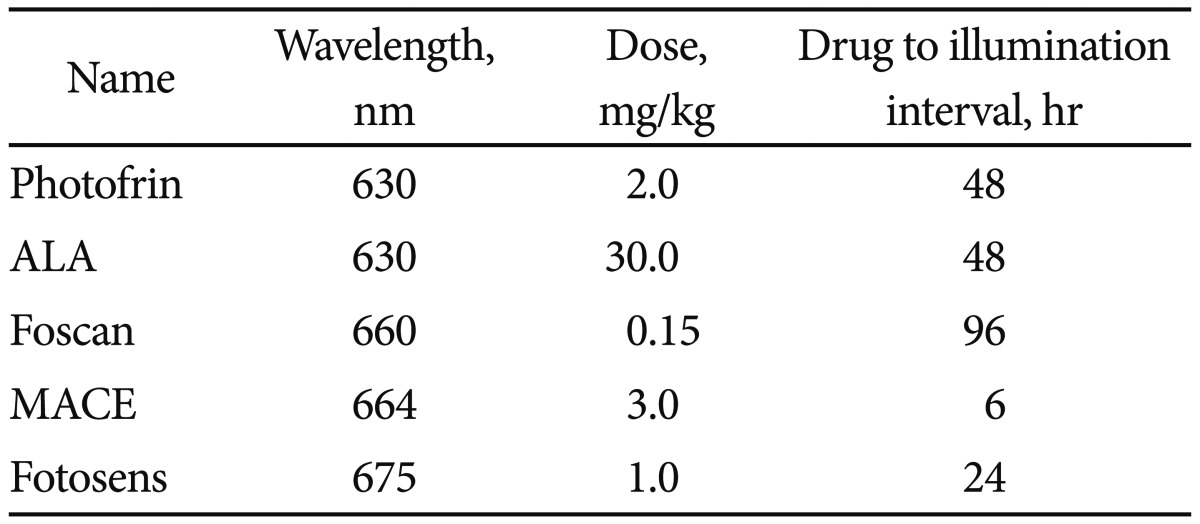
PS agents are natural or synthetic structures that transfer light energy.6 As photosynthesis is a form of light transfer that is the basis of life on Earth, it should come as no surprise that many structures allow for light transfer. However, to be considered a successful PS agent for PDT, the structure must allow for the specific creation of the PDR, which is detailed later in this section. Not surprisingly, the chlorophyll derivatives from plants and bacteria are excellent PS agents. So too are dyes and porphyrins. While thousands upon thousands of structures have been identified as PS agents, perhaps two dozen have been well characterized and only about one dozen employed in clinical trials. Of these perhaps two or three are Food and Drug Administration (or equivalent) approved and commercially available. In the clinic, a successful PS agent has most of the following characteristics: nontoxic till activated, hydrophilic for easy systemic application, activated by a clinically useful light wavelength (see below), and reliable generation of the PDR. It also concentrates in tumor, clears normal tissue, is eliminated from the patient relatively rapidly, is a nontoxic degradation product with ease of synthesis, pain free therapy and, just as importantly, commercial availability. Several excellent PS agents cannot be used simply because they are unavailable for purchase. Many countries have developed PSs that are approved for clinical use in their own territory and which have proved to be perfectly safe but are not licensed to be administered in another country. Currently several PS agents are on the market and while each creates the PDR they are not interchangeable. It is worth a brief review of the benefits and shortcomings of each. Please see Table 1.
Hematoporphyrin derivative (HPD)
HPD (Photofrin; Axcan Pharma/Pinnacle Inc., Mont-Saint-Hilaire, Canada) was the first commercially available drug and has the most clinical experience. This is the PS brought to market by Dougherty in the 1970's. HPD is a mixture of various monomers, dimmers, and polymers of hematoporphyrin, all required for successful PDT. This PS is nontoxic and upon activation with light does not cause pain, allowing for easy and reliable outpatient treatment. The drug is not an active producer of the PDR so treatment times can be 20 minutes or more per lesion. Further the drug is retained in various organs, including the skin for 6 to 8 weeks post introduction. Unintentional exposure of skin to sunlight (a strong light source) during this time frame can cause unintentional PDT to exposed skin. This can result in severe burn.
M-tetrahydroxophenyl chlorine (mTHPC) (Foscan)
This plant based chlorine derivative is synthetically pure and produces a rapid and significant PDR. Treatment time is measured in seconds. The drug is so active that after infusion patients must stay in a dark room for 24 hours as room light will activate the drug and cause a severe burn (dark toxicity). In addition, the treatment is painful so most individuals undergo illumination while under anesthesia. Still, as it is highly effective, the drug has found a niche in the treatment of primary and recurrent head and neck cancers.
Mono-L-aspartyl chlorine e6 (NPe6)
Marketed under different names such as MACE, LS11, NPe6, this derivative is called Fotolon (RUE Belmedpreparaty, Minsk, Republic of Belarus). This plant based chlorine is a very effective agent to generate the PDR. It does not have the dark toxicity common with Foscan (Biolitec Pharma Ltd., Dublin, Ireland) and also allows treatment several hours after infusion. In contrast to Photofrin or Foscan, this PS allows for same day infusion and therapy which is very convenient for patients and practitioners.
Aminolevulinic acid (ALA)
ALA is a prodrug that is enzymatically converted to Proto-Porphyrin IX, a potent PS. ALA can be topically placed, orally introduced or intravenously injected. Whilst the treatment is uncomfortable when topically placed, no systemic phototoxicity occurs. As such, ALA treatment has found a niche for cutaneous disease.
Fotosens
This is a dye based PS that can be activated shortly after infusion. This allows for same day therapy in a single sitting. However, illumination can take 20 minutes per lesion and several weeks of skin photosensitivity is to be expected.
Illumination
Each PS has a unique wavelength of light and intensity of light fluence required for successful activation. Clinically, red light -630 nm can penetrate tissue to perhaps 0.5 cm which allows for both surface and more deep seated tumor illumination.6,7 Some PS may activate at higher wavelength for deeper penetration, others at lower wavelengths for more superficial illumination. By choosing the appropriate PS and light wavelength, one can optimize treatment. For example, a superficial skin lesion may be illuminated with blue light -400 nm which allows only a 1 mm light penetration.6-9 When using topical ALA, PDT ablates the skin lesion but spares deep normal tissue of the skin. Clinically, this translates into successful cancer therapy without scarring of normal tissue. ALA, HPD, MACE, and Foscan can activate at multiple light wavelength from blue to green to red, again allowing for more selective illumination depth based on the individual tumor's depth and location of surrounding critical structures.6 This is an underutilized characteristic of these PS which could be better exploited in the clinic.
The light source itself can vary tremendously.7 As long as light of the appropriate wavelength and fluence activates the PS, successful PDT can occur. Intense visible multispectrum light from a lamp or precise laser light (tuned to the PSs) are currently successful light sources for PDT. Currently, regulatory agency approved PS also have regulatory agency approved light sources which are commercially available. As laser remains relatively expensive, though highly reliable; new light sources such as light emitting diodes (LED's) have been developed (Quantel Ltd., Newbury, UK). These are generally very small and portable, so new treatment paradigms such as prolonged or repeated (metronomic) PDT are now possible.
As the light itself must be brought to the region of treatment, a variety of procedures for PDT have been developed to accomplish this.8 Fiberoptic cables or the LED itself can be brought to the tumor bed by direct placement during surgery or endoscopy. In addition, ultrasound or computed tomography guided placement is also possible, similar to the brachytherapy procedure used in radiation oncology. With current technology, light sources can be reliably introduced into virtually all of human anatomy.
PDR
When light of the appropriate wavelength and intensity bathes the PS, a PDR may occur.9 Fundamentally, specific wavelength light activates the PS which then leads to a series of photochemical reactions that ideally allow for tumor destruction without undue normal tissue injury. Light energy alters the inert PS via electron transfer from the photon of light to the PS. This activated PS may then lose energy by several pathways. Energy loss may occur by release of light. This fluorescent phenomenon, may be observed, and allows for detection and delineation of tumor. Fluorescence detection is an underutilized pathway of the PS; it can assist in targeting tumor and defining normal tissue borders (which do not fluoresce) since minimal PS is in surrounding normal tissue. The active PS can also lose energy by creating a type I photochemical reaction. This Fenton reaction creates free radicals which are destructive. However, the most important pathway for clinical PDT is the generation of a type II photochemical reaction which is termed the PDR. Here the PS interacts with oxygen to generate singlet oxygen, which is considered to be the basis of PDT's tumor and vascular ablation ability. This oxygen dependent type II PDR is a sine qua non for PDT. The half life of singlet oxygen is in the order of 40 nanoseconds which allows for destruction of a radius of 20 nanometers.10 Whilst a truly tiny volume, clearly it is enough for clinical success.
PDT MECHANISMS
How exactly the PDR translates into successful clinical outcomes is a work in progress.11 As described, singlet oxygen has a radius of destruction measured in nanometers yet this allows for a significant and complex cascade of events resulting in local, regional, and systemic alteration of both tumor and immune response so that reliable tumor control is possible. This section will describe the mechanisms of action of PDT arbitrarily separated as cellular events, vascular events and systemic immune events. In vivo, these will occur simultaneously or nearly so.
Tumor cell
Tumor destruction from PDT can occur by both programmed (apoptotic) pathways and non-programmed (necrosis) pathways.12,13 This is fortunate as some tumors have developed genetic mutations eliminating or minimizing apoptosis.13 On a cellular and sub cellular level, the PS is brought to the malignancy through various mechanisms including:14-16 receptor mediated phagocytosis/endocytosis, low density lipoprotein receptor binding, lipid binding, uptake via tyrosine kinase/epidermal growth factor receptor, diffusion, biodistribution and perhaps many other yet undiscovered pathways. Each PS may have a preferred method of uptake into tumor cells.17,18 As an example Photofrin, composed of multiple sized porphyrins, is taken up by multiple pathways and is concentrated in not only the cell membrane but also various organelle membranes such as mitochondria. In contrast, MACE, which is amphiphilic, concentrates in mitochondria. Foscan appears to concentrate in the golgi apparatus and endoplasmic reticulum.
Generally, when high light intensity is employed, the tumor cells are rapidly ablated by necrosis.19 The cellular and sub cellular membrane destruction is rapid. Most probably, calcium20 and metabolic byproducts21 are released which are not compatible with cell function and repair functions are overwhelmed, leading to ablation of the tumor cell. This also leads to release of cytokines and toxic chemicals from, for example, the mitochondria.22 This leakage will then create lethal damage in cells nearby23 (bystander effect) as well as creating a regional and systematic reaction, which is described below.
In contrast, apoptotic death may be initiated by PDT, generally when low light doses are employed.24 During apoptosis, the cells cease to function and undergo an orderly and programmed dissolution. No bystander effect or immune response is expected as no toxic chemicals are leaked. Apoptotic pathways are found in both tumor and normal cells across many species including bacteria. It appears that apoptosis is a well conserved method of the organism to eliminate damaged cells. PDT appears to be able to activate this pathway.
It should be noted that PS is believed to preferentially concentrate in the rapidly dividing cells of malignancy whilst clearing preferentially from surrounding normal tissue which retains little PS. Therefore, ideally the PDR is lethal to tumors without affecting normal tissue. In reality, any cell with PS that is activated may undergo necrosis or apoptosis and, if normal tissue containing significant amounts of PS are exposed to light, severe tissue morbidity can be expected (i.e., skin photosensitivity).6
Vascular events
Just as in tumor cells, endothelial cells of the vascular systems can concentrate PS.17,18,25 This may be due to similar mechanisms including receptors, diffusion, and multiple other pathways, including some yet to be described. The neovasculature of malignancy is also felt to be leaky due to poor and incomplete cellular borders and may serve as an additional means for PS accumulation in tumor regions as it leaks through. In contrast, the surrounding normal vasculature in the nontumorous region may facilitate PS clearance.
Just as in tumor, PS in vasculature, when activated by appropriate light, will create the PDR. Several events will occur. By disrupting the vascular walls, blood will not flow to the tumor and oxygen will become scarce.26 When this occurs, necrosis is to be expected both of the involved neo vasculature and nearby tumor cells. The release of toxic chemicals, excess calcium and other intra cellular debris will lead to blockage and collapse of the micro vasculature feeding the tumor. Platelets will be activated and aggregate.27 Overall, a rapid loss of blood supply in concert with direct tumor and vascular cell lysis will be a lethal event to the tumor. Also possible, is a less intense reaction where light penetrates at lower fluences. Here, apoptosis pathways of the vasculature may occur, again leading to tumor hypoxia and destruction but without cytokine and immune activation. Clinically, both apoptosis and necrosis occurs in the neovasculature contributing significantly to tumor cell destruction not only directly by lack of blood and hypoxia but also by release of toxic substances such as thromboxanes, platelet aggregators, and various toxic cytokines which will also prime the immune system.28 Potentially, depending on the clinical situation, one may be able to illuminate in such a fashion as to favor apoptotic versus necrotic pathways.
Immune system
PDT may enhance immune response and surveillance.29 It is probable that long-term tumor control is a combination of direct PDT effects on the lesion and its vasculature in combination with up regulation of the immune system. When PDT induces necrosis of tumors and their vasculature, an immune cascade is also initiated.30 Release of inflammatory mediators occurs from the treated region, which include various cytokines, growth factors and proteins. This release stimulates various white blood cells to be activated including neutrophils and macrophages which converge on the treatment region. It is felt that significant tumor cell death occurs from these activated immune cells.31,32 Upon arrival, macrophages phagocytize PDT damaged cancer cells and present proteins from these tumors to CD4 helper T lymphocytes, which then activate CD8 cytotoxic T lymphocytes. Not only does this immune reaction occur at the PDT site, it also may occur at regional and distant lymphatic tissue. These cytotoxic T cells may not only cause necrosis but may also induce apoptotic pathways whenever tumor cells are found, even after PDT is complete.
Clinically, patients treated with PDT show elevated levels of various cytokines and histologic evaluations of treated tumors routinely show immune cell infiltration, again pointing to the immunomodulatory effect of PDT. Much work needs to be done to better define and manipulate the immune system response of PDT in particular and cancer in general.
CLINICAL APPLICATION OF MECHANISMS
In the clinical situation, application of PDT is translated into a two step procedure. In the first, presensitisation, a PS is administered systemically, by intravenous injection, or topically. In the second, illumination, the presensitised tissue is exposed to a specific light whose wavelength matches the absorption band of the chemical/drug. The ensuing PDR is presented as necrotic destruction of the tissue under treatment.
Clinicians have far more opportunity to improve PDT than simply pointing the light at the target. As described, PDT is an interplay of drug and light both of which can be manipulated to maximize outcome in various clinical situations. Again, for simplicity, we will describe modifications and outcomes for drug dose, light dose and PDR separately.
Drug dose
PS accumulates in the malignant region and is cleared from normal tissue. One strong means of maximising PDT in tumor and sparing normal tissue is to use as little PS as possible to achieve tumor response.33 When using a clinically determined minimal concentration of PS, the tumor will still concentrate enough PS for an affective PDR, while normal tissue, which has less PS, will not be damaged. The standardised dose of PS may not always be the optimal dose.
Light
Light localization is critical for optimal and accurate PDT; however, localisation of the tumor bed may not always be obvious. By employing fluorescence of the PS, the clinician can often visualize tumor.34 This helps define a more precise therapy. Furthermore, loss of fluorescence post-PDT therapy may indicate tumor ablation far better than a preprogrammed time of illumination. The use of fluorescence both for detection of disease and treatment outcome is underutilized. Intense illumination has been the basis of clinical PDT; this was historically due to difficulties in getting the light source to the lesion. The advantage of high fluence is that more light photons penetrate deeply allowing a more complete illumination at depth. When using low PS drug dose, high light fluence allows for excellent response rates without undue morbidity. In contrast, high drug dose with high light fluence rates create normal tissue damage as significant PDR can occur in normal tissue that has been flooded by PS. High light doses also push toward necrotic pathways which may have implications for immune response. Low light fluences allow for apoptosis and less inflammation and immune response. This may have clinical ramifications. For example, intense therapy in a tight space, such as the airway or esophagus may lead to abundant normal tissue reaction and obstruction due to necrosis and inflammation. Lower light doses might prevent this.
Drug light intervals (DLI)
When PS is first introduced it is travelling in the vascular supply. With time, PS accumulates in tumor and is less likely to be in the blood. So the DLI can be a critical component of controlling PDT.35 By illuminating early after PS introduction, vascular effects would predominate; later illumination would favor tumor cell effect. Potentially prolonged DLI could give a more select tumor destroying effect.36 In contrast, rapid DLI would favor vascular collapse. In a highly vascular tumor, this may be preferential.37
PDR
By altering drug dose, light dose and DLI,38,39 clinicians may provide for necrotic pathway PDT, apoptotic pathway PDT, selectively vascular destructive PDT, selectively tumor destructive PDT and even immunomodulatory PDT. Through rationally designed clinical trials that further explore these varied treatment paradigms, PDT will be able to advance dramatically as an oncologic intervention.
CONCLUSIONS
PDR results from the interaction between a chemical PS and a specific wavelength of light in the presence of oxygen. The interaction releases cytotoxic species, notably singlet oxygen. The overall mechanism involves molecular, subcellular and vascular changes which bring about necrosis and/or apoptosis of the tumor. The parameters of the individual components of PDT, such as the PS, light and the interval between presensitization and illumination, can be modulated to achieve cellular (direct) or vascular response predominance. We are still far from achieving optimal objectives. For now, even with our current understanding of the mechanism of PDT, the clinician has a double edge therapeutic sword to target and inflict injuries to the tumor. On the one hand, the operator can localize the lesion visually or by imaging to direct the light to it. On the other hand, by the very fact that the PS is accumulated in the tumor tissue and that it attracts its own specific wavelength of light, the tumor will be specifically targeted, provided that the light reaches the presensitized tissue. This is akin to the situation where the indentation of a key matches that of the lock allowing its unlocking.