INTRODUCTION
In 1958, Caroli was the first to describe congenital dilatation of the intrahepatic bile ducts (IHDs), which leads to bile stasis and stone formation [1]. The disorder can involve all IHDs or may be limited to some part of the liver, usually the left lobe or a segment of the left lobe [2,3]. Its incidence is extremely low (1 in 1,000,000 of the population) [2], and it comprises two entities: the pure form, or Caroli disease (CD), and a form associated with periportal fibrosis, also known as the Caroli syndrome [4]. The disease is the consequence of the arrest or derangement of the normal embryologic remodeling of bile ducts, and results in varying degrees of destructive inflammation and segmental dilatation [5]. Congenital hepatic fibrosis, CD, and Caroli syndrome are generally considered a disease continuum at different stages of severity associated with ductal plate malformation [6]. CD is the result of the involvement of large IHDs, while abnormal development of the small interlobular bile ducts results in congenital hepatic fibrosis. If all levels of the biliary tree are affected, features of both congenital hepatic fibrosis and CD are expressed as the Caroli syndrome [5]. The main clinical manifestations are intermittent abdominal pain, hepatomegaly, recurrent cholangitis, and stone formation.
We report a case of simple-type CD with diffuse liver involvement, which was diagnosed in adulthood. Our case demonstrates a distinctively characteristic central dot sign with multiple stones in the IHDs and the common bile duct (CBD) via imaging studies. CBD stones were treated successfully with endoscopic retrograde cholangiopancreatography (ERCP).
CASE REPORT
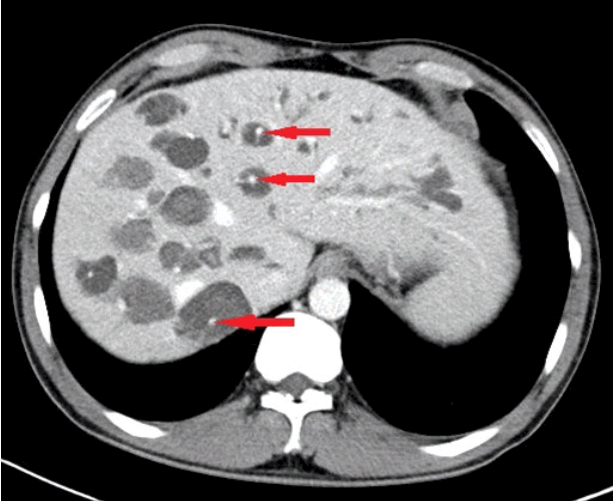
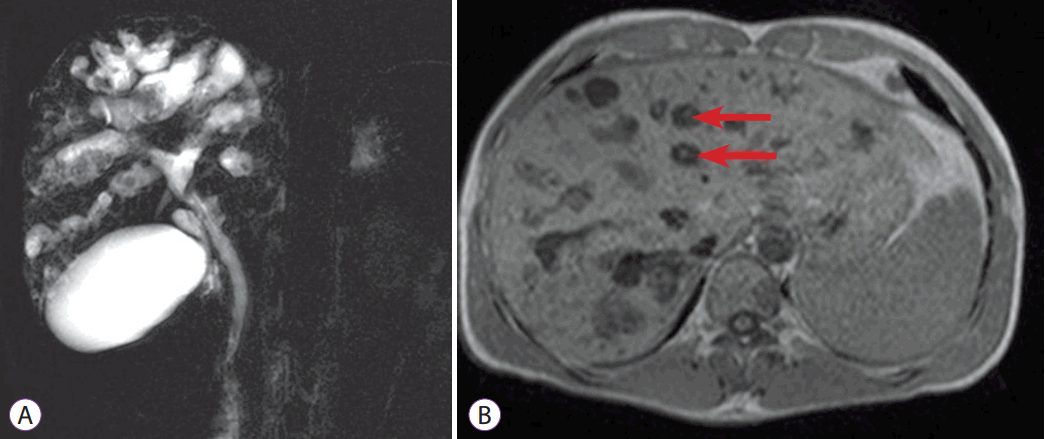
A 42-year-old man was admitted with an one-month history of intermittent fever and right upper quadrant abdominal pain. One year prior to admission, he visited our outpatient clinic because of an incidental finding of elevated alkaline phosphatase (ALP) and gamma glutamyl transferase (GGT). He had no known underlying disease or significant family history, and denied alcohol and tobacco consumption. At that time, jaundice, fever, or stigmata of chronic liver disease were not observed and no signs of portal hypertension were noted. Abdominal computed tomography (CT) (Fig. 1) and magnetic resonance cholangiopancreatography (MRCP) (Fig. 2) showed multiple saccular dilatations of both IHDs with typical central dot signs and inner stone formation. Hepatic involvement was more marked in the right lobe, leading to the diagnosis of diffuse-type CD. There was no evidence of associated polycystic kidney disease by imaging studies. Since the patient had no specific symptoms or signs, he was followed up regularly with a prescription for ursodeoxycholic acid (UDCA), and remained asymptomatic until admission.
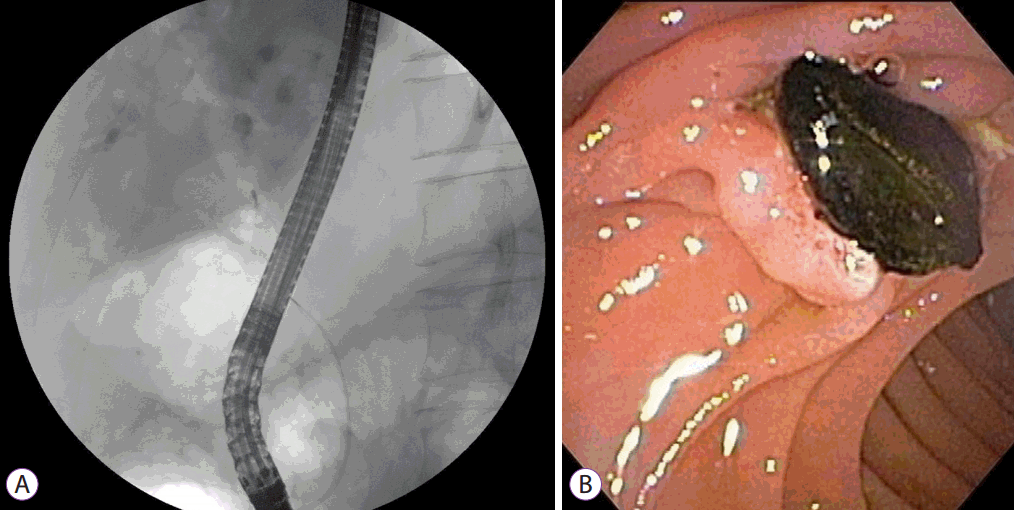
On admission, vital sign measurements were as follows: blood pressure, 100/80 mm Hg; body temperature, 38.0┬░C; pulse rate, 100 beats/minute; and respiratory rate, 20 breaths/minute. The physical examination showed right upper quadrant tenderness and laboratory test results were as follows: white blood cell count, 10,192/mL; aspartate transaminase, 33 IU/L; alanine transaminase, 41 IU/L; ALP, 882 IU/L; GGT, 531 IU/L; total bilirubin, 0.52 mg/dL; and cancer antigen 19-9, 34.3 U/mL. Other laboratory indicators of portal hypertension, such as platelet count, serum albumin, or prothrombin time were all within normal range. Abdominal CT again showed multiple saccular dilatations of the IHD with inner stone formation, and mild dilatation of the biliary tree with distal CBD stones. ERCP showed several CBD stones and cystic dilatation of both IHDs, which were communicating with the biliary tree (Fig. 3A). Black pigment CBD stones were removed using the basket method after endoscopic sphincterotomy (Fig. 3B). The symptoms subsided and the patient was discharged from the hospital with no procedure-related complications. After 2 years, he remains in clinical follow-up, with no other complaint.
DISCUSSION
CD is a rare congenital disorder characterized by communicating cavernous ectasia or segmental cystic dilatation of the IHD. Although duct abnormalities are generally present at birth, patients are often asymptomatic until adolescence or adulthood [3], as in our case. Mode of inheritance is unclear, but in most cases, CD is transmitted in an autosomal recessive fashion [4]. Two forms of CD have been described. The simple or isolated type is characterized by cystic dilatation of the IHD, while the more common complex variantŌĆöalso known as Caroli syndromeŌĆöis associated with congenital hepatic fibrosis, portal hypertension, and polycystic kidney disease [7-9]. Simple-type CD is often limited to one lobe (usually the left), whereas Caroli syndrome usually involves the entire liver [10]. Our case was diagnosed as simple-type CD with diffuse liver involvement, which is uncommon, because there was no evidence of portal hypertension or polycystic kidney disease.
Febrile episodes caused by cholangitis are the most common symptoms in CD; however, this may not be accompanied by jaundice or abdominal pain. Hepatomegaly is usually present, and laboratory tests typically show an elevated level of ALP and GGT. Intrahepatic ductal dilatation induces bile stasis, which may predispose individuals to stone formation and frequent infections, such as cholangitis, abscess formation, or even septicemia. Patients with CD are also at risk for intrahepatic cholangiocarcinoma [11]. It is reported that about 7% of patients eventually develop malignancy [12] and the risk is known to be as high as 100-fold greater in patients with CD than in the general population [13,14].
The diagnosis of CD relies on demonstrating the cystic dilatation of IHD in continuity with the biliary tree by imaging studies. The central dot sign is suggested as a pathognomonic finding of CD. These are small foci of strong contrast enhancement within cystic lesions and are detected by CT, magnetic resonance imaging, or on an ultrasonogram. These correspond to fibrovascular bundles containing a portal vein radicle and a branch of the hepatic artery, bridging the saccule, and they appear as central dots or a linear streak [15]. MRCP and ERCP are valuable in identifying direct communication of the intrahepatic saccular dilatation with the biliary tree [16]. However, because of its invasiveness, ERCP is recommended for therapeutic purposes but not for the diagnosis of CD. In this case, ERCP was performed for treatment of choledocholithiasis, and direct communication of an intrahepatic cyst with the bile duct was confirmed.
Clinicians should also be aware of the differential diagnosis of CD, such as the von Meyenburg complex, a rare condition that is usually asymptomatic and does not cause liver function abnormalities. It is generally diagnosed incidentally by MRCP, showing multiple small-sized cystic nodules (<1.5 cm) that do not communicate with the biliary tree [17]. Other differential diagnosis of CD include primary sclerosing cholangitis, recurrent pyogenic cholangitis, polycystic liver disease, and biliary papillomatosis [5].
Treatment of CD depends on the clinical symptoms and location of the biliary abnormalities. In most patients, UDCA is used in an attempt to reduce the formation of intrahepatic stones [18]. In our case, CBD stones removed during ERCP were pigment stones; therefore, it is reasonable to assume that IHD stones are also pigment stones, so that a successful result is less expected with UDCA. Therapeutic ERCP with dilatation of strictures and extraction of stones is also helpful in some cases. Potential curative treatment of CD is surgical resection, such as segmentectomy, lobectomy, or hepaticojejunostomy, determined by the range of distribution [19]. In localized diseases, lobectomy can not only cure but also reduce the risk of subsequent occurrence of cholangiocarcinoma [10]. Patients with end-stage liver disease or diffuse-type CD, as in our case, should be considered for eventual liver transplantation [10,20].
In conclusion, this patient had a sporadic form of simple-type CD with diffuse involvement of the liver, and was diagnosed in adulthood following incidental discovery of liver function test abnormalities. The characteristic central dot sign was clearly observed on imaging studies. The patient had coexisting IHD stones and experienced abdominal pain associated with a CBD stone which was successfully treated with ERCP. Since our patient showed diffuse involvement of both liver lobes, the possibility of developing portal hypertension should be considered and regular follow-up is needed. In case of cirrhosis or cholangiocarcinoma, liver transplantation should be considered. Although CD is a rare congenital disorder, it must be recognized and included in the differential diagnosis of patients with nonspecific abdominal pain, recurrent cholangitis, or chronic cholestasis of unknown cause.